
We are building and applying advanced genomic and computational technologies to study viral pathogens.
 We use genomics to gain insights into the emergence, spread, and evolution of outbreaks. Over the past decade, we’ve tackled viral pathogens such as Ebola, Lassa, Zika, Mumps, and SARS-CoV-2, while fostering collaborative research programs in Nigeria, Sierra Leone, and Senegal. We support frontline responders and public health efforts through technology transfer and training. We are working to expand our genomic surveillance and epidemiology efforts, integrating sequencing with diagnostics and advanced analytics, and we’re creating a hub for diagnostics, sequencing, and analytics to support local and global public health initiatives.
We use genomics to gain insights into the emergence, spread, and evolution of outbreaks. Over the past decade, we’ve tackled viral pathogens such as Ebola, Lassa, Zika, Mumps, and SARS-CoV-2, while fostering collaborative research programs in Nigeria, Sierra Leone, and Senegal. We support frontline responders and public health efforts through technology transfer and training. We are working to expand our genomic surveillance and epidemiology efforts, integrating sequencing with diagnostics and advanced analytics, and we’re creating a hub for diagnostics, sequencing, and analytics to support local and global public health initiatives.
Gire et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014. | Metsky et al. Zika virus evolution and spread in the Americas. Nature 2017 | Siddle et al. Transmission from vaccinated individuals in a large SARS-CoV-2 Delta variant outbreak. Cell 2021
 To track emerging pathogen threats in real time, we are developing a new class of diagnostics that leverage CRISPR-Cas technologies for enhanced accuracy and sensitivity combined. We are developing new machine learning methods for rapid, optimized diagnostic design across all diagnostic modalities. We are also developing experimental and computational techniques for unbiased deep sequencing-based detection of viruses
To track emerging pathogen threats in real time, we are developing a new class of diagnostics that leverage CRISPR-Cas technologies for enhanced accuracy and sensitivity combined. We are developing new machine learning methods for rapid, optimized diagnostic design across all diagnostic modalities. We are also developing experimental and computational techniques for unbiased deep sequencing-based detection of viruses During the 2014 West African Ebola outbreak and COVID-19, we showed how viral mutations impact the unfolding of an outbreak. We collaborated with the Jeremy Luban lab to show that mutations in receptor proteins conferred increased infectivity in human cells. We have worked with partners to conduct single-cell profiling of Ebola and other BL-4 pathogens to investigate details of gene regulation and immune responses. We have explored the therapeutic potential of CRISPR-Cas systems, showing their success in reducing viral load and infectivity in mammalian cells. We have created massively parallel technologies to identify the wide range of peptides viruses produce and studying which of these they present to our immune systems.
During the 2014 West African Ebola outbreak and COVID-19, we showed how viral mutations impact the unfolding of an outbreak. We collaborated with the Jeremy Luban lab to show that mutations in receptor proteins conferred increased infectivity in human cells. We have worked with partners to conduct single-cell profiling of Ebola and other BL-4 pathogens to investigate details of gene regulation and immune responses. We have explored the therapeutic potential of CRISPR-Cas systems, showing their success in reducing viral load and infectivity in mammalian cells. We have created massively parallel technologies to identify the wide range of peptides viruses produce and studying which of these they present to our immune systems. 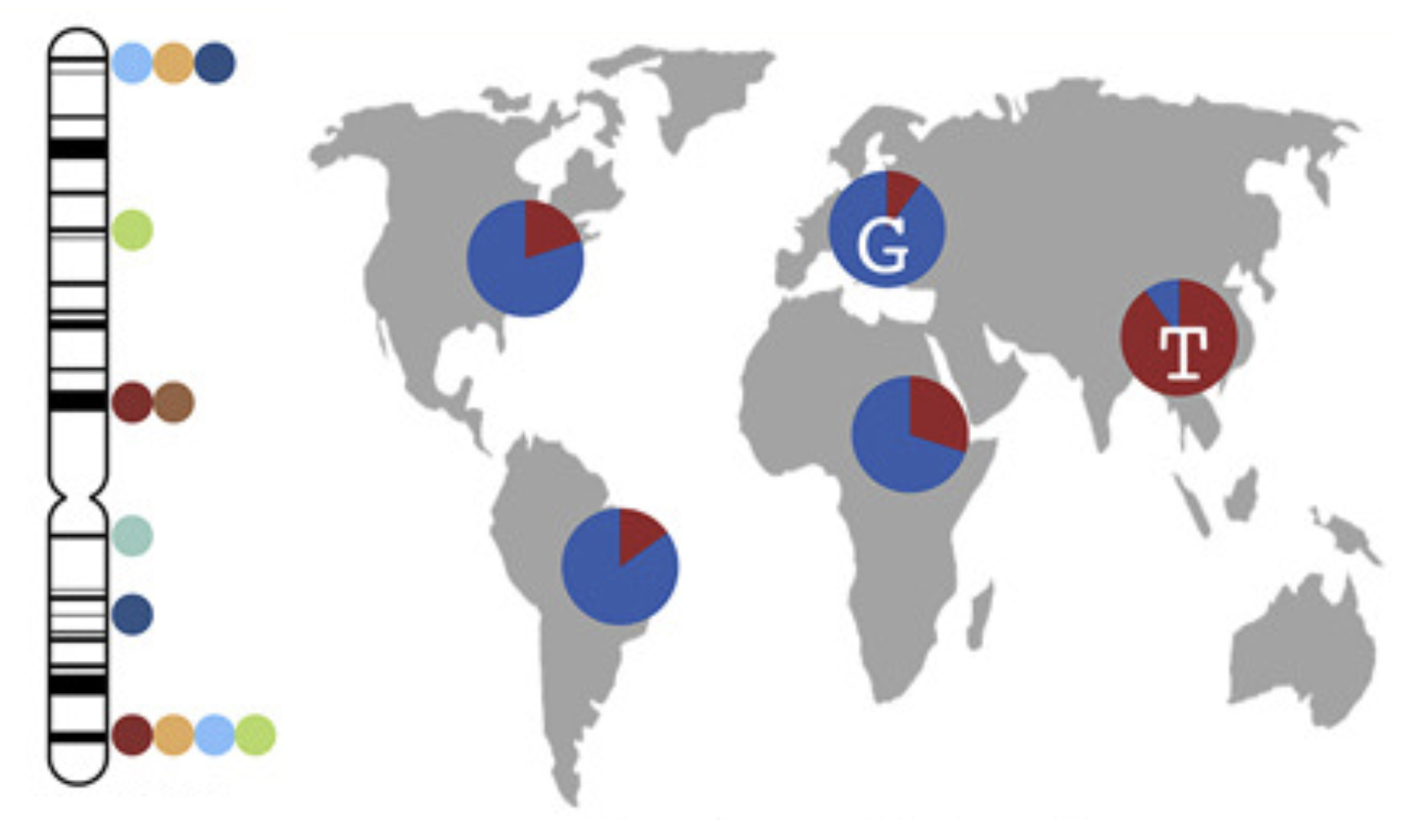 Most disease-associated and evolutionarily adaptive mutations occur in non-coding regions, which are difficult to study through conventional means. We developed a massively parallel reporter assay to investigate differences in expression between alleles at thousands of sites in non-coding regions. Our lab has leveraged our cross-disciplinary work in human disease and viral evolution, to discover viral capsids that target specific cell types for precise gene delivery. Finally, we continue to study human evolution around the world, characterizing background patterns of variation, developing neutral demographic models and simulations, and constructing machine learning approaches to localize signals of selection.
Most disease-associated and evolutionarily adaptive mutations occur in non-coding regions, which are difficult to study through conventional means. We developed a massively parallel reporter assay to investigate differences in expression between alleles at thousands of sites in non-coding regions. Our lab has leveraged our cross-disciplinary work in human disease and viral evolution, to discover viral capsids that target specific cell types for precise gene delivery. Finally, we continue to study human evolution around the world, characterizing background patterns of variation, developing neutral demographic models and simulations, and constructing machine learning approaches to localize signals of selection.